|
REACCIÓN DEL TEJIDO NEURAL AL DAÑO.
La degeneración axonal es un proceso que ocurre como consecuencia
de una amplia variedad de trastornos metabólicos, tóxicos,
hereditarios e inflamatorios. Patologías como esclerosis múltiple,
esclerosis lateral amiotrófica y neuropatías periféricas
tienen como consecuencia la degeneración axonal. El mecanismo
por el cual se inicia el proceso de degeneración walleriana es
motivo de constante estudio, sugiriéndose un mecanismo de respuesta
al daño similar a la apoptosis (1).
Dentro de los primeros minutos de producida la lesión, la membrana
plasmática del axón se cierra. En el segmento proximal
se acumula contenido axonal (neurofilamentos, vesículas, mitocondrias)
debido a la interrupción del flujo axoplásmico. El segmento
distal al sitio de la lesión degenera (2).
Los fenómenos más tempranos de la degeneración
walleriana se visualizan a las 24 horas de producida la lesión.
El evento crucial en la degeneración del segmento distal del
axón lesionado es la desintegración granular del citoesqueleto
y el axoplasma. La lesión estructural del axón se correlaciona
con la pérdida de la capacidad para generar y conducir el potencial
de acción (3). La desintegración del citoesqueleto es
gatillada por un aumento en la concentración de calcio axoplásmico
(4). Se ha postulado que el aumento del calcio libre intraaxonal se
relaciona con la patogenia del daño axonal en trauma (5, 6) e
isquemia (7,8). Estos estudios sugieren la participación de los
canales de calcio voltaje-sensibles (VSCC) y del canal intercambiador
de Na+/Ca2+ en su fisiopatología.
En el segmento distal de la lesión, la pérdida del contacto
de las células de Schwann con los axones produce una disminución
de los niveles de RNAm de proteínas componentes de la mielina,
producidos por la glía. Estas proteínas son la glicoproteína
asociada a mielina, la proteína cero y la proteína básica
de la mielina. Las células de Schwann se desdiferencian y proliferan
con aumento de receptores de factor de crecimiento neural, de proteína
acídica fibrilar glial factor de maduración glial-b ,
molécula de adhesión L1 y molécula de adhesión
neural (9).
PAPEL DEL CALCIO EN LA DEGENERACIÓN AXONAL.
La entrada de calcio es un componente necesario para el inicio de la
degeneración axonal. La degeneración walleriana no ocurre
si la concentración intracelular de calcio libre es menor de
200 mM (5). El aumento del nivel de calcio en el citoplasma activa proteasas
calcio-dependiente, las cuales digieren los neurofilamentos, parte importante
del citoesqueleto del axón (10,11).
La transección del axón produce entrada de calcio (12,13,14).
Diferentes estudios han demostrado que axones dañados mantenidos
en un medio con quelante de calcio degeneran más lentamente que
los axones en un medio con alta concentración de calcio, con
la consiguiente activación de proteasas axonales calcio dependientes
(15). También es posible inhibir la degeneración axonal
al sustituir el calcio del medio extracelular por cobalto o manganeso,
iones que no activan las proteasas calcio-dependientes (16,17).
El mecanismo por el cual los niveles de calcio aumentan en el axoplasma
no han sido bien establecidos, pero existen evidencias que apoyan la
participación de VSCC axonales en este proceso (18,19).
MECANISMOS DE HOMEOSTASIS DEL CALCIO EN NEURONAS.
Las neuronas utilizan el calcio en el control del crecimiento celular
y diferenciación (20), mantención de la estructura del
citoesqueleto (21), excitabilidad de la membrana (22), exocitosis y
actividad sináptica (23).
En neuronas no depolarizadas la concentración de calcio intracelular
libre ([Ca2+]i) es 100 nM, mientras que la concentración
de calcio en el espacio extracelular es 2 mM. La gradiente de calcio
se mantiene por la interacción de cuatro categorías de
eventos: entrada, tamponamiento, almacenamiento en depósitos
intracelulares y salida activa de calcio libre al espacio extracelular
(24).
Al producirse daño axonal aumenta el flujo de calcio al axolema
a través de mecanismos de trasporte calcio-específicos
como canales de sodio gatillados por voltaje (25), intercambiador Na+/Ca2+
(26) y VSCC (5,8). Se ha observado protección contra la degeneración
axonal al bloquear VSCC tipo N en conjunto con VSCC tipo L. Los autores
han sugerido una baja densidad de VSCC tipo N en relación a VSCC
tipo L, por lo que el flujo de calcio en la degeneración estaría
dado principalmente por VSCC tipo L. Estos resultados podrían
estar influidos por la participación de VSCC tipo T, P/Q o R.
Los VSCC se encuentran en la membrana plasmática de células
musculares, neuronas y células gliales (27). Estos canales regulan
la entrada de calcio a la célula, mediando funciones fisiológicas,
tales como, crecimiento neuronal, regulación enzimática,
expresión génica y salida de neurotrasmisores de los terminales
nerviosos. Los VSCC se clasifican según sus propiedades electrofisiológicas
y farmacológicas en tipos L,T, N, P/Q y R (28).
La activación de los VSCC depende de su localización
subcelular y del potencial de membrana local. Por ejemplo, en neuronas
piramidales de hipocampo, los canales tipo L se concentran en la base
de las dendritas apicales, mientras que los canales tipo N se encuentran
ampliamente distribuidos (29).
También existen canales de calcio activados por ligandos. Los
receptores ionotrópicos son sensibles a agonistas como NMDA y
kainato, los cuales inducen su apertura. La activación de los
canales NMDA ha sido asociada a fenómenos de plasticidad sináptica
y muerte celular (30).
Los receptores metabotrópicos median la acción de los
canales iónicos por mecanismos dependientes de proteínas
que unen GTP, con formación de IP3 y DAG, lo cual
moviliza calcio de depósitos intracelulares.
Los bajos niveles de calcio intracelular se mantienen gracias a la
acción de proteínas capaces de unir calcio libre como
calbindina y parvalbúmina. La capacidad de tamponamiento de las
proteínas es limitada. Se ha podido establecer que en neuronas
existen mecanismos de secuestro de calcio libre intracelular en organelos
(31). Estos incluyen el retículo endoplásmico liso, mitocondrias
y vesículas sinápticas (32).
En las neuronas existen diversos mecanismos que permiten eliminar calcio
citosólico al extracelular y mantener los niveles de 100 nM contra
la gradiente electroquímica. Dentro de estos mecanismos se encuentra
el intercambiador Na+/Ca+2 y la bomba de calcio
dependiente de ATP, las cuales sacan calcio de la célula o lo
almacenan en compartimentos intracelulares, como retículo endoplásmico
liso y mitocondrias. El intercambiador de Na+/Ca+2
saca calcio desde el citoplasma al extracelular y lo intercambia por
Na+, utilizando la gradiente química del sodio sin
consumir ATP (33).
Al aumentar la concentración de calcio libre citoplasmático,
éste se une a la calmodulina. La calmodulina se une a enzimas
como las calmodulina-quinasa y adenilato ciclasa, activándolas.
También se activan las calpaínas, las cuales digieren
los neurofilamentos del citoesqueleto, por lo que se fragmenta el axolema
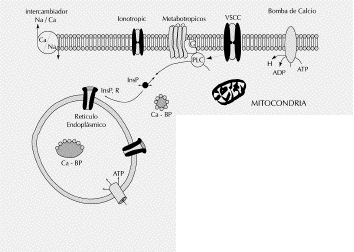
e inicia la degeneración axonal (fig.1)
|
|
|
Figura 1. MECANISMOS REGULADORES DE LA CONCENTRACIÓN
DE CALCIO EN EL ESPACIO INTRACELULAR. Los VSCC forman parte
de los mecanismos de homeostasis del calcio neuronal. Una vez
activados, entra calcio al espacio intracelular. Si los mecanismos
de tamponamiento (calbindina y calmodulina) y salida (intercambiador
Na/Ca y bomba ATP dependiente) son superados, se activan proteínas
quinasa, fosfolipasa C (PLC) y proteasas calcio-dependientes tipo
calpaínas. Uno de los efectos observados es la digestión
de los neurofilamentos, lo cual inicia el proceso de degeneración
axonal. InsP: inositol fosfato, InsP-R: receptor de inositol fosfato.
Ca-BP: proteína unidora de calcio.
|
CANALES DE CALCIO VOLTAJE-SENSIBLES.
Los VSCC son proteínas compuestas por una subunidad a
1, la cual forma el poro, y unidades accesorias a
2/d y b. Se han identificado seis
subunidades a1 diferentes (A, B, C, D, E y S) (34).
Canal de calcio tipo L.
El canal tipo L expresado en el sistema nervioso es activado por
potenciales de alto voltaje. Está formado por una subunidad
a 1D que forma el poro y las subunidades
a 2- d, b y g regulatorias y de anclaje. Las subunidades a1,
b y g son codificadas por genes separados, mientras que las subunidades
a 2 y d son producto de la proteolisis de un precursor mayor codificado
por un solo gen (35). Este canal es bloqueado por 1-4 dihidropiridina
(DHP), por fenilalquilamina, y por diltiazem (36).
La subunidad a 1 contiene el sitio de
unión de las dihidropiridinas (DHP), moléculas que bloquean
la apertura del canal. La secuencia aminoacídica primaria es
consistente con una proteína de transmembrana con cinco sitios
potenciales de N-glicosilación y sitios de potencial fosforilación
dependiente de AMPc.
La subunidad a 2 tiene un peso molecular
de 165-220 kDa, el cual disminuye a 130-150 kDa después de
la reducción de puentes disúlfuros que la unen con la
subunidad g, de 24-33 kDa (37).
La subunidad b tiene un peso de 52-65
kDa. Es un substrato para la proteína quinasa dependiente de
AMPc y PKC. Esta unidad se encuentra fuertemente unida a la subunidad
a1 y se ha relacionado con la regulación de la actividad del
canal tipo L.
La subunidad g del receptor de DHP es
una glicoproteína de 30-33 kDa. La subunidad d es de 24-33 kDa. Esta subunidad tiene un domino hidrofóbico
transmembrana y se encuentra unida por puentes disulfuros a la subunidad
a2. Se desconocen sus funciones dentro
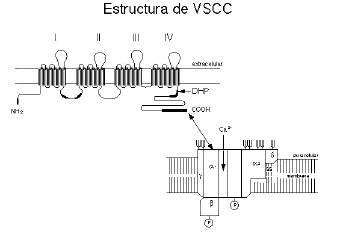
del canal. (fig.2)
|
|
|
Figura 2. ESTRUCTURA DE LOS CANALES DE CALCIO VOLTAJE-SENSIBLES.
Esquema superior: estructura de subunidad a1. Esta subunidad
está formada por cuatro dominios homólogos (I-IV),
que contienen seis regiones transmembrana. Los VSCC tipo L presentan
un dominio que une dihidropiridina (DHP) en el extremo C-terminal
de la zona IV. El esquema inferior representa la interacción
entre las subunidades de VSCC, con sitios de glicosilación
en el espacio extracelular y sitios de fosforilación
(P) en el espacio intracelular.
|
Canal de calcio tipo N.
El canal de calcio N es activado por potenciales de alto voltaje.
Este canal es insensible a DHP. Ha sido descrito sólo en células
de origen neuronal y su expresión se encuentra aumentada en
células PC12 (línea celular derivada de feocromocitoma
de rata) tratadas con factor de crecimiento neural. El canal de calcio
N participa en la excitabilidad de membrana, crecimiento axonal, migración
neuronal (38) y salida de neurotransmisores (39). Este canal es bloqueado
selectivamente por -conotoxina GVIA (40). Consiste en una subunidad
1B de 230 kDa, a2/d de 150 KDa, b3 de 57 kDa y un
polipéptido de 94 kDa que no se encuentra en el canal tipo
L (41).
En modelos de encefalomielitis autoinmune y esclerosis múltiple
se ha encontrado acumulación e integración a la membrana
plasmática de subunidad 1B de VSCC tipo N en axones en degeneración.
Esta distribución de los canales de calcio podría influir
en un aumento del flujo de calcio al axoplasma, contribuyendo al proceso
de degeneración axonal (19). Los VSCC tipo L han sido demostrados
en los cuerpos celulares de neuronas de ganglios espinales, pero no
existe evidencia clara sobre su expresión en membranas axonales.
CONCLUSIONES.
Los VSCC se encuentran ampliamente distribuidos en diferentes tipos
celulares excitables y no excitables. Corresponden a la principal vía
de entrada de calcio en el músculo cardiaco y el músculo
liso, participan también en la salida de neurotransmisores desde
células endocrinas y neuronas sensitivas, y en procesos de degeneración
axonal (5). Por lo tanto estrategias que permitan proteger contra la
degeneración axonal inducida por axotomía podrían
hacerlo también en neuropatías periféricas. En
este sentido existe evidencia que apoya que la inhibición de
calpaínas es un elemento protector contra la degeneración
axonal por exposición a vincristina (42).
La degeneración axonal es un proceso activo modulable en variadas
etapas relacionadas con la homeostasis del calcio intracelular y ,por
lo tanto, un blanco interesante para explorar nuevos tratamientos para
enfermedades que presentan degeneración axonal tanto en el sistema
nervioso central como periférico.
REFERENCIAS.
- Finn J, Weil M, Archer F, Siman R, Srinivasan A, Raff M. Evidence
that wallerian degeneration and localized axon degeneration induced
by local neurotrophin deprivation do not involve caspases. J. Neurosci.
20, 1333-1341, (2000).
- Waller A. Experiments on the section of the glossopharryngeal and
hypoglossal nerves of the frog and observations of the alterations produced
thereby in the structure of their primitive fibers. Phil. Trans. R.
Soc. Lond. (Biol) 140, 423-429. (1850)
- Oblinger M, Lasek R. Axotomy-induced alterations in yhe synthesis
and transport of neurofilaments and microtubules in dorsal root ganglion
cells. J. Neurosci. 8, 1747-1758. (1988)
- Schlaepfer W. Structural altarations of peripheral nerve induced by
the calcium ionophore A23187. Brain Res. 136, 1-9. (1977)
- George E, Glass J, Griffin J. Axotomy-induced axonal degeneration
is mediated by calcium influx through ion-specific channels. J. Neurosci.
15, 6445-6454. (1995)
- A, Siman R, Trojanowski J, Povlishock J. The role of calpain-mediated
spectrin proteolysis in traumatically induced axonal injury. J. Neuropathol.
Exp. Neurol. 58, 365-375. (1999)
- Stys P, Waxman S, Ransom B. Na-Ca exchanger mediates Ca influx during
anoxia in mammalian central nervous system white matter. Ann. Neurol.
30, 375-380. (1991)
- Fern R, Ransom B, Waxman S. Voltage-gated calcium channels in CNS
white matter: role in anoxic injury. J. Neurophysiol. 74, 369-377. (1995)
- Martini R, Bollensen E, Schachner M. Immunocytological localization
of the major peripheral nervous system glycoprotein P0 and the L2/HNK-1
and L3 carbohydrate structures in developing and adult mouse sciatic
nerve. Dev Biol. 129, 330-338. (1988)
- Schlaepfer W, Bunge R. The effects of calcium ion concentration on
the degradation of amputed axons in tissue culture. J. Cell Biol. 59,
456-470. (1973)
- Nixon R, Quackenbeish R, Vitto A. Multiple calcium-activated neural
proteases (CANP) in mouse retinal ganglion cell neurons: specificities
for endogenes sbstrates and comparison to purified brain CANP. J. Neurosci.
6, 1252-1263. (1986)
- Lopachin R, Lopachin V, Saubermann A. Effects of axotomy on distribution
and concentration of elements in rat sciatic nerve. J Neurochem 54,
320-332. (1990)
- Xie X, Barret J. Membrane releasing in cultured rat septal neurons
after neurite transection: evidence for enhancement by Ca tiggered protease
activity and cytoskeletal disassembly. J. Neurosci. 11, 3257-3267. (1991)
- Ziv N, Spira M. Spatiotemporal distribution of Ca following axotomy
and throughout the recovery process of cultured Aplysia neurons. Eur.
J. Neurosci. 5, 657-668. (1993)
- Schlaepfer W, Lee C, Lee V, Zimmerman U. An immunoblot study of neurofilament
degradation in situ and during calcium-activated proteolysis. J. Neurochem.
44, 502-509. (1985)
- Zimmerman U, Schlaepfer W. Characterization of a brain calcium-activated
protease that degrades neurofilament proteins. Biochemistry 21, 3977-3983.
(1982)
- Mehdi S. Cell-penetrating inhibitors of calpain. Trends Biochem. Sci.
16, 150-153. (1991)
- DiPolo R, Caputo C, Bezanilla F Voltage-dependent calcium channel
in the squid axon. Proc. Natl. Acad. Sci. USA 80, 1743-1745. (1983)
- Kornek B, Storch M, Djamshidian A, Weissert R, Wallstroem E, Stefferl
A, Zimprich F, Olsson T, Linington C, Schmidbauer M, Lassmann. Distribution
of a calcium channel subunit in dystrophic axons in multiple sclerosis
and experimental autoimmune encephalomyelitis. Brain 12, 1114-1124.
(2001)
- Yuste R, Katz L. Control of postsynaptic calcium influx in developing
neocortex by exitatory and inhibitory neurotransmitters. Neuron 6, 333-344.
(1991)
- Trifaro J, Vitale M Cytoskeleton dynamics during neurotransmitter
release. Trends Neurosci. 16, 466-474 (1993)
- Marty A, Neher E. Potassium Channels in cultured bovine adrenal chromaffin
cells. J.Physiol .367, 117-141. (1985)
- Robitaille R, Charlton M. Presynaptic calcium signals and transmitter
release are modulated by calcium-activated potassium channels. J. Neurosci.
297-305. (1992)
- Tymianski M, Tator C. The direct effect of nimodipine on intracellular
calcium in spinal neurons in explant cultures. Drugs Dev. 2, 337-347.
(1993)
- Stys P, Lopachin R Mechanisms of calcium and sodium fluxes in anoxic
myelinated central nervous system axons. Neuroscience 82, 21-32.(1998)
- Stys P, Waxman S, Ransom B. Ionic mechanisms of axoxic injury in mammalian
CNS white matter: role of Na channels and Na-Ca exchanger. J. Neurosci.
12, 430-439. (1992)
- Powers P, Liu S, Hogan K, Gregg R. Skeletal muscle and brain isoforms
of a b-subunit of human voltage-dependent calcium channels are encoded
by a single gene. J. Biol. Chem. 267, 22967-22972. (1992)
- Puro D, Hwang J, Kwon O, Chin H. Characterization of an L-type calcium
channel expressed by human retinal Müller (glial) cells. Mol. Brain
Res. 37, 41-48. (1996)
- Westenbroek R, Hell J, Warner C, Dubel S, Snutch T, Caterall W. Biochemical
properties and subcellular distribution of an N-type calcium channel
alpha 1 subunit. Neuron 9, 1099-1115 (1992)
- Bliss T, Collergridge G. A synaptic model of memory: long-term potentiation
in the hippocampus. Nature 361, 31-39. (1993)
- Kaplin A, Ferris C, Voglmaier S, Smyner S. Purified reconstituted
inositol, 1,4,5, triphosphate receptors. Thiol reagents act directly
on receptor preotein. J. Biol. Chem. 269, 28972-28978. (1994)
- Werth J, Thayer S. Mitochrondria buffer physiological calcium loads
in cultured rat dorsal root ganglion neurons. J. Neurosci. 14, 348-356
(1994)
- Blaustein M. The effect of cyanide on the efflux of calcium from squid
axons.J. Physiol. 200, 497-527 (1969).
- Snutch T, Reiner P. Ca2+ channels: diversity of form and function.
Curr. Opin. Neurobiol. 2, 247-253. (1992)
- Catterall W. Functional subunit structure of voltage-gated calcium
channels. Science 253, 1499-1500. (1991)
- Hofmann F, Oeken H, Schneider T, Sieber M. The biochemical properties
of L-type calcium channels. J. Cardiovasc. Pharmacol. 12, 525-530. (1988)
- Takahashi M, Seagar M, Jones J, Reber B, Catterall W. Subunit structure
of DHP-sensitive calcium channels from skeletal muscle. Proc. Natl.
Acad. Sci USA 84, 5478-5482. (1987)
- Komuro H, Rakic P. Role of N-type calcium channels in neuronal migration
Science 257,806-809. (1992)
- Hirning L, Fox A, McCleskey E, Olivera B, Thayer S, Miller R, Tsien
R. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine
from sympathetic neurons Science 239, 57-61. (1988)
- Olivera B, Cruz L, de Santos V, Griffin D, Gray W, Rivier J. Neural
calcium channel antagonists. Discrimination between calcium channel
subtypes using _-conotocin from Conus magus venom. Biochemistry 26,
2086-2090. (1987)
- Whitcher D, De Waard M, Sakamato J, Franzini C, Pragnell M, Kahl S,
Campbell K. Subunit identification and reconstitution of the N-type
Ca2+ channel complex purified from brain. Science 261, 486-489. (1993)
- Wang M, Wu Y, Culver D, Glass J. Pathogenesis of axonal degeneration:
parallels between wallerian degeneration and vincristine neurophaty.
Neuropathol. Exp. Neurol. 59, 599-606. (2000)
|


